









Contents [show]
Sindrom Treacher-Collins
Penyakit genetik yang jarang berlaku, sindrom Teacher-Collins dicirikan oleh perkembangan kecacatan kelahiran tengkorak dan muka semasa hayat embrio, mengakibatkan kecacatan pada muka, telinga dan mata. Akibat estetik dan fungsi adalah lebih atau kurang teruk dan sesetengah kes memerlukan banyak campur tangan pembedahan. Walau bagaimanapun, dalam kebanyakan kes, mengambil alih membolehkan kualiti hidup tertentu dipelihara.
Apakah Sindrom Treacher-Collins?
definisi
Sindrom Treacher-Collins (dinamakan sempena Edward Treacher Collins, yang pertama kali menggambarkannya pada tahun 1900) adalah penyakit kongenital yang jarang berlaku yang menunjukkan dirinya sejak lahir dengan kecacatan yang lebih atau kurang teruk pada bahagian bawah badan. muka, mata dan telinga. Serangan adalah dua hala dan simetri.
Sindrom ini juga dipanggil sindrom Franceschetti-Klein atau mandibulo-facial dysostosis tanpa kelainan hujung.
Punca
Tiga gen setakat ini diketahui terlibat dalam sindrom ini:
- gen TCOF1, terletak pada kromosom 5,
- gen POLR1C dan POLR1D, masing-masing terletak pada kromosom 6 dan 13.
Gen ini mengarahkan pengeluaran protein yang memainkan peranan penting dalam perkembangan embrio struktur muka. Perubahan mereka melalui mutasi mengganggu perkembangan struktur tulang (terutamanya pada rahang bawah dan atas serta tulang pipi) dan tisu lembut (otot dan kulit) bahagian bawah muka semasa bulan kedua kehamilan. Pinna, saluran telinga serta struktur telinga tengah (ossicles dan / atau gegendang telinga) juga terjejas.
Diagnostik
Kecacatan muka boleh disyaki daripada ultrasound pada trimester kedua kehamilan, terutamanya dalam kes kecacatan telinga yang ketara. Dalam kes ini, diagnosis pranatal akan diwujudkan oleh pasukan pelbagai disiplin daripada pengimejan resonans magnetik (MRI) janin, membolehkan kecacatan divisualisasikan dengan lebih tepat.
Selalunya, diagnosis dibuat melalui pemeriksaan fizikal yang dilakukan semasa lahir atau tidak lama kemudian. Oleh kerana kebolehubahan besar kecacatan, ia mesti disahkan di pusat khusus. Ujian genetik pada sampel darah mungkin diarahkan untuk mencari keabnormalan genetik yang terlibat.
Sesetengah bentuk ringan tidak disedari atau akan dikesan secara kebetulan lewat, contohnya berikutan kemunculan kes baharu dalam keluarga.
Sebaik sahaja diagnosis dibuat, kanak-kanak itu tertakluk kepada beberapa siri pemeriksaan tambahan:
- pengimejan muka (x-ray, imbasan CT dan MRI),
- pemeriksaan telinga dan ujian pendengaran,
- penilaian penglihatan,
- cari apnea tidur (polysomnography)…
Orang yang berkenaan
Sindrom Treacher-Collins dianggap menjejaskan satu daripada 50 bayi yang baru lahir, kedua-dua perempuan dan lelaki. Dianggarkan kira-kira 000 kes baharu muncul setiap tahun di Perancis.
Faktor-faktor risiko
Kaunseling genetik di pusat rujukan disyorkan untuk menilai risiko penghantaran genetik.
Kira-kira 60% kes muncul secara berasingan: kanak-kanak itu adalah pesakit pertama dalam keluarga. Kecacatan berlaku berikutan kemalangan genetik yang menjejaskan satu atau lain sel pembiakan yang terlibat dalam persenyawaan (“mutasi de novo”). Gen yang bermutasi kemudiannya akan diturunkan kepada keturunannya, tetapi tidak ada risiko khusus untuk adik-beradiknya. Walau bagaimanapun, ia harus diperiksa sama ada salah seorang daripada ibu bapanya sebenarnya tidak mengalami bentuk sindrom kecil dan membawa mutasi tanpa mengetahuinya.
Dalam kes lain, penyakit ini adalah keturunan. Selalunya, risiko penghantaran adalah satu dalam dua dengan setiap kehamilan, tetapi bergantung kepada mutasi yang terlibat, terdapat cara penghantaran lain.
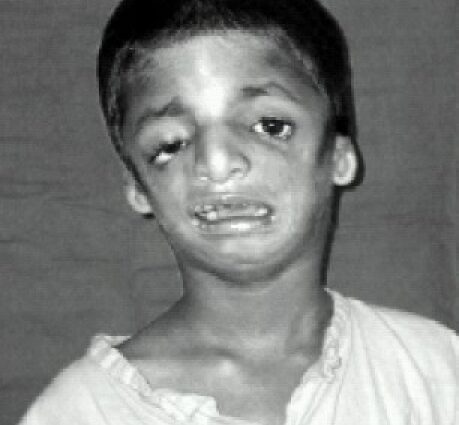
Gejala sindrom Treacher-Collins
Ciri-ciri wajah mereka yang terjejas selalunya adalah ciri, dengan dagu yang atrofi dan surut, tulang pipi yang tidak wujud, mata condong ke bawah ke arah pelipis, telinga dengan pavilion yang kecil dan berkelim teruk, atau bahkan tidak hadir sama sekali …
Gejala utama dikaitkan dengan kecacatan sfera ENT:
Kesukaran pernafasan
Ramai kanak-kanak dilahirkan dengan saluran pernafasan atas yang sempit dan mulut terbuka sempit, dengan rongga mulut kecil sebahagian besarnya terhalang oleh lidah. Oleh itu, kesukaran bernafas yang ketara terutamanya pada bayi baru lahir dan bayi, yang dinyatakan dengan berdengkur, apnea tidur dan pernafasan terlalu lemah.
Kesukaran makan
Pada bayi, penyusuan susu ibu mungkin terhalang oleh kesukaran bernafas dan oleh kelainan pada lelangit dan lelangit lembut, kadangkala terbelah. Memberi makan lebih mudah selepas pengenalan makanan pepejal, tetapi mengunyah boleh menjadi sukar dan masalah pergigian adalah perkara biasa.
Pekak
Ketidakselesaan pendengaran akibat kecacatan telinga luar atau tengah terdapat dalam 30 hingga 50% kes.
Gangguan visual
Satu pertiga daripada kanak-kanak mengalami strabismus. Ada juga yang mungkin rabun dekat, hiperopik atau astigmatik.
Kesukaran pembelajaran dan komunikasi
Sindrom Treacher-Collins tidak menyebabkan defisit intelek, tetapi pekak, masalah penglihatan, kesukaran pertuturan, kesan psikologi penyakit serta gangguan yang disebabkan oleh rawatan perubatan yang sering sangat berat boleh menyebabkan kelewatan. bahasa dan kesukaran berkomunikasi.
Rawatan untuk sindrom Treacher-Collins
Penjagaan bayi
Sokongan pernafasan dan/atau pemberian tiub mungkin diperlukan untuk memudahkan pernafasan dan penyusuan bayi, kadangkala sejak lahir. Apabila bantuan pernafasan mesti dikekalkan dari masa ke masa, trakeotomi (pembukaan kecil di trakea, di leher) dilakukan untuk memperkenalkan kanula secara langsung memastikan laluan udara dalam saluran udara.
Rawatan pembedahan kecacatan
Intervensi pembedahan yang lebih atau kurang kompleks dan banyak, berkaitan dengan lelangit lembut, rahang, dagu, telinga, kelopak mata dan hidung boleh dicadangkan untuk memudahkan makan, pernafasan atau pendengaran, tetapi juga untuk mengurangkan kesan estetik kecacatan.
Sebagai petunjuk, celah lelangit lembut ditutup sebelum umur 6 bulan, prosedur kosmetik pertama pada kelopak mata dan tulang pipi dari 2 tahun, pemanjangan mandibula (gangguan mandibula) ke arah usia 6 atau 7 tahun, pemasangan semula pinna telinga pada usia sekitar 8 tahun, pembesaran saluran pendengaran dan / atau pembedahan ossicles sekitar 10 hingga 12 tahun… Pembedahan pembedahan kosmetik lain masih boleh dilakukan pada masa remaja…
Bantuan pendengaran
Alat bantu pendengaran kadangkala boleh digunakan dari umur 3 atau 4 bulan apabila pekak menjejaskan kedua-dua telinga. Pelbagai jenis prostesis boleh didapati bergantung pada sifat kerosakan, dengan kecekapan yang baik.
Tindakan susulan perubatan dan paramedik
Untuk mengehadkan dan mencegah hilang upaya, pemantauan berkala adalah pelbagai disiplin dan memanggil pelbagai pakar:
- ENT (risiko tinggi jangkitan)
- Pakar oftalmologi (pembetulan gangguan penglihatan) dan pakar orthoptist (pemulihan mata)
- Doktor gigi dan ortodontik
- Jurupulih Pertuturan…
Sokongan psikologi dan pendidikan selalunya diperlukan.